BIOVIA DISCOVERY STUDIO 2022 cracked suite
$ 150.00
BIOVIA modeling and simulation software allows scientists to perform computations of chemical, biological and materials properties; to simulate, visualize and analyze chemical and biological systems; and to communicate the results to other scientists. Scientists can investigate and test hypotheses in silico prior to costly experimentation, reducing the time and expense involved in bringing products to market.
BIOVIA Discovery Studio life science modeling and simulation application supports in silico target identification and lead optimization using a wealth of trusted life science modeling and simulation methods.
The industry-leading BIOVIA scientific software portfolio integrates the diversity of science, experimental processes and information requirements. It connects people, processes and data end-to-end, across research, development, QA/QC and manufacturing.
Capabilities include Scientific Informatics, Molecular Modeling & Simulation, Data Science, Laboratory Informatics, Formulation Design, BioPharma Quality & Compliance and Manufacturing Analytics
Description

Comprehensive Modeling & Simulation for the Life Sciences
Today’s biopharmaceutical industry is marked by complexity: growing market demands for improved specificity and safety, novel treatment classes and more intricate mechanisms of disease. Keeping up with this complexity requires a deeper understanding of therapeutic behavior.
Modeling and simulation methods provide a unique means to explore biological and physicochemical processes down to the atomic level. This can guide physical experimentation, accelerating the discovery and development process.
BIOVIA Discovery Studio brings together over 30 years of peer-reviewed research and world-class in silico techniques such as molecular mechanics, free energy calculations, biotherapeutics developability and more into a common environment. It provides researchers with a complete toolset to explore the nuances of protein chemistry and catalyze discovery of small and large molecule therapeutics from Target ID to Lead Optimization.
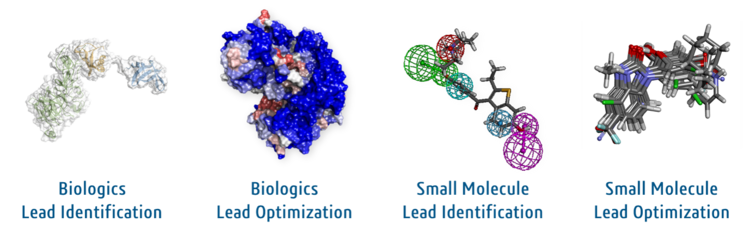
With Discovery Studio you can:
- Investigate and test hypotheses in silico prior to costly experimental implementation, thus reducing the time and expense involved in bringing products to market
- Drive scientific exploration from target identification to lead optimization with a wealth of trusted life science modeling and simulation tools
- Leverage BIOVIA Pipeline Pilot to automate processes, create and deploy custom workflows, and integrate data types, databases, and third-party or in-house tools
- Enhance personal productivity and boost team collaboration by enabling researchers to share data and make better informed decisions
Search
- Perform multiple sequence searches using BLAST and PSI-BLAST against local or NCBI databases
- For multiple chain proteins, simultaneously and independently perform multiple sequence alignments of each protein chain
- Predict the transmembrane helices in transmembrane protein sequences
- Predict sites prone to Post Translational Modifications (PTMs) using sequence-based motif searching
Model
- Analyze and prepare structures from 3D structure repositories (e.g., PDB)
- Generate 3D structure models using MODELER
- Verify the quality of a structure model
- Use LOOPER to systematically search loop conformations and rank using CHARMm
- Graft loop conformations from a template structure onto a target model
- Systematically optimize amino acid side-chains using ChiRotor CHARMm simulations
- Use ZDOCK to perform protein-protein docking and examine binding partner interactions
- Study conformational flexibility with explicit solvent-based Molecular Dynamics (MD) simulations using CHARMm or NAMD
Design
- Predict electrical properties of the protein, including pH-dependent stability and protonation states and the isoelectric point
- Conduct thermal or pH-based mutational stability and binding affinity predictions
- Identify potential stable disulfide bridge locations
- Calculate biophysical properties important for protein formulation, including viscosity and solubility
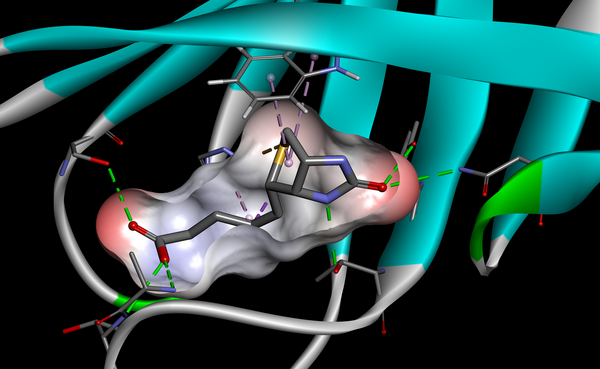
Prepare
- Analyze and prepare 3D structures (e.g., PDB, X-ray structure, homology model) for SBD
- Automatically build neighboring molecules based on crystal packing and analyze their interactions
- Predict residue ionization states at chosen pH
- Identify and study putative ligand binding sites
- Prepare ligands with extensive set of characteristics and calculate 3D coordinates
- Generate ligand conformations
- Filter ligands based on drug-likeness, molecular properties, or to remove undesirable groups or features
Screen
- Hit Identification and optimization
- Perform virtual screening on ligands and fragments using either the CATALYST pharmacophore engine, or the LibDock or CDOCKER docking approaches
- Perform docking with GOLD §
- Perform in situ lead optimization using classical medicinal chemistry reaction transformations and commercially-available reagents
- Scaffold-hop or perform R-group substitutions in situ using molecular fragments derived from commercially-available compounds
Score
- Calculate binding energies with MM-PBSA or MM-GBSA CHARMm-based methods
- Accurately predict relative ligand binding energy for a congeneric ligand series using the free energy perturbation (FEP) method
- Calculate the relative free energy of binding for a combinatorial library of ligands modeled by Multi-Site Lambda Dynamics (MSLD)
- Identify critical interacting residues using a comprehensive set of favorable, unfavorable and unsatisfied non-bond monitors
- Profile and prioritize screening hits, optimizing potency and target specificity
Extend
- Design and optimize combinatorial libraries as new starting points for further screening.
- Combine your scores with classical QSAR, fingerprints, and Quantum Mechanics based descriptors and create advanced predictive models
- Minimize toxicity using TOPKAT and optimize the pharmacokinetic profile.
https://youtu.be/tZIC1FIvnm8?si=xn2gnh0n2mwfZHAz
Build
- Automatically generate pharmacophores from the data available
- Sets of active ligands
- Receptor binding sites
- Receptor-ligand complexes
- Perform rigorous Pharmacophore validation based on sets of control compounds with known activity.
- Hypotheses can include
- Geometric, feature-based queries
- Shape-similarity
- “Forbidden” space
- Go beyond the limitations of classical pharmacophore elucidation algorithms by exploring Ensemble Pharmacophores for very large/diverse compound sets with a risk of multiple modes of action
Apply
Conduct robust Pharmacophore screening studies
- Build and search databases of 3D conformations
- Consider and analyze the full conformational space of your ligands
- Explore off-target activity and drug repurposing using the PharmaDB* database
Discovery Studio now includes the most extensive reported database for ligand profiling. Built from, and validated using, the scPDB*, the PharmaDB contains approximately 240,000 receptor-ligand pharmacophore models.
Design
Design and characterize your ligands and combinatorial libraries
- Enumerate reaction- or core-based libraries
- Enumerate ionization states, tautomers and isomers
- Filter poor candidates with undesirable functional groups and Lipinski and Veber rules or your own criteria
- Calculate numerous physicochemical and fingerprint properties
- Optimize combinatorial libraries using Pareto optimization, diversity and similarity analysis
- Clustering tools and 3D visualization using PCA analysis
Understanding and quantifying structure-activity relationships can significantly impact lead optimization and drug development by minimizing tedious and costly experimentation.
Build, validate and apply your own models based on a wide range of approaches, and keep improving them as new data become available.
Assess the potential risk posed by unfavorable pharmacokinetic properties and potential toxicity using BIOVIA’s distributed models, extend them to better cover your proprietary chemical space and use the comprehensible indications to navigate around such liabilities.
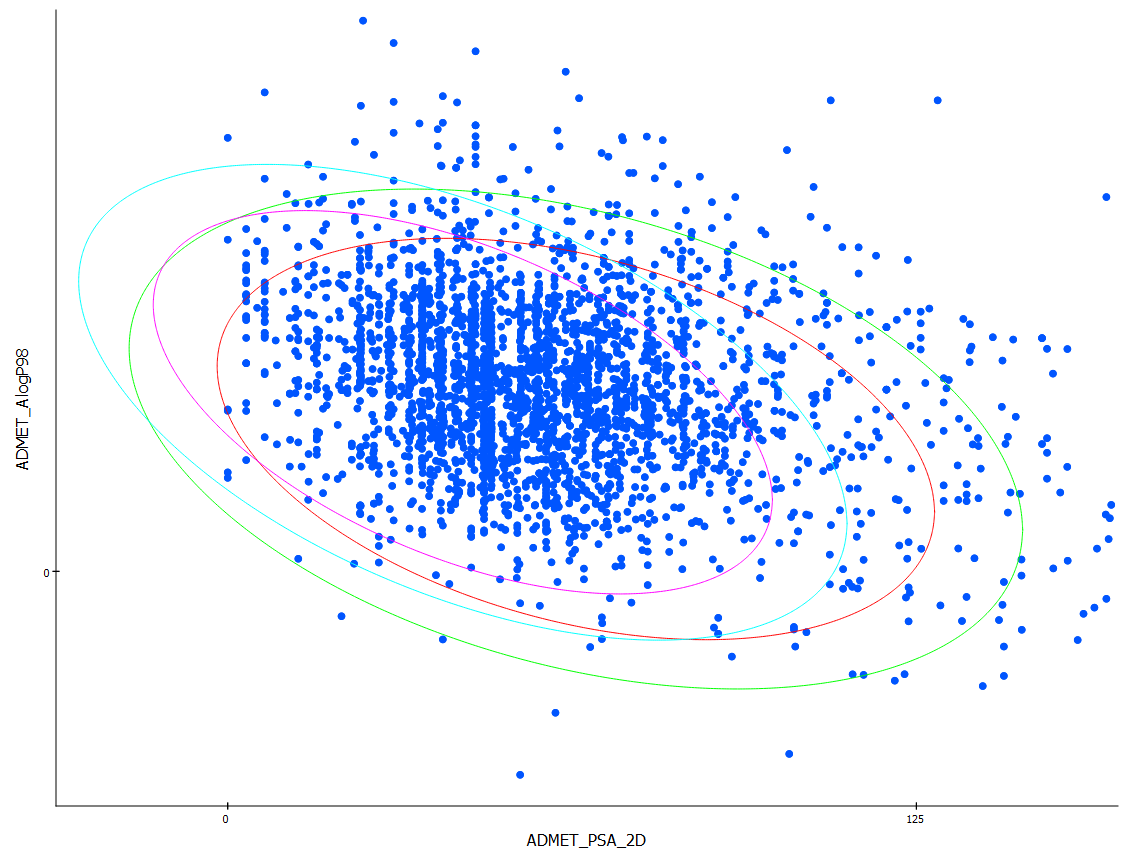
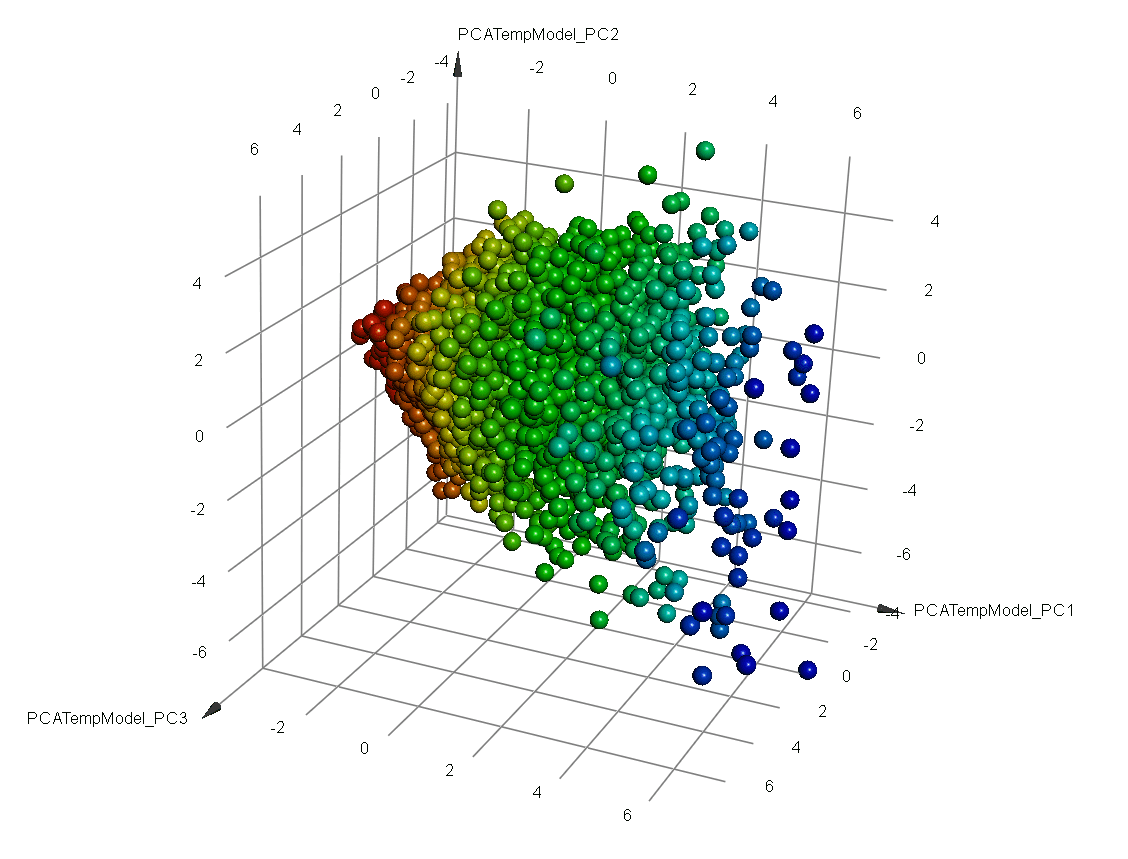
QSAR
- Comprehensive and consistent data preparation:
- Ligands: remove duplicates and handle tautomers and ionization
- Prepare response property (scaling and binning)
- Split your data into training and test sets with the appropriate methods
- Choose from a large number of physicochemical, topological, electronic, geometric. fingerprint and Quantum Mechanics based descriptors
- Create statistical models including Bayesian, MLR (Multiple Linear Regression), PLS (Partial Least Squares), and GFA (Genetic Functional Analysis)
- Analyze and validate models using model applicability domains (MAD), automatic test set validation, cross validation and statistical metrics
- Identify Matched Molecular Pairs (MMPs) transformations and study activity cliffs
ADMET
Get an early assessment of your compounds by calculating the predicted ADMET (absorption, distribution, metabolism, excretion and toxicity) properties for collections of molecules such as synthesis candidates, vendor libraries, and screening collections. Use the results to eliminate compounds with unfavorable ADMET characteristics and evaluate proposed structural refinements, designed to improve these properties prior to synthesis.
ADMET descriptors include:
- Human intestinal absorption
- Aqueous solubility
- Blood brain barrier penetration
- Plasma protein binding
- CYP2D6 binding
- Hepatotoxicity
- Filter sets of small molecules for undesirable function groups based on published SMARTS rules
Toxicity
Evaluate your compounds’ performance in experimental assays and animal models. Compute and validate assessments of the toxic and environmental effects of chemicals solely from their molecular structure. TOPKAT® (TOxicity Prediction by Komputer Assisted Technology) employs robust and cross-validated Quantitative Structure Toxicity Relationship (QSTR) models for assessing various endpoints and utilizing the patented Optimal Predictive Space validation method to assist in interpreting the results.
Please note:
Details on our extensible TopKat models have been published in QSAR Model Report Format (QMRF) on the European Commission Joint Research Center (JRC)’s “QSAR Model Database”*.
- Ames mutagenicity
- Rodent carcinogenicity (NTP and FDA data)
- Weight of evidence carcinogenicity
- Carcinogenic potency TD50
- Developmental toxicity potential
- Rat oral LD50
- Rat maximum tolerated dose
- Rat inhalation toxicity LC50
- Rat chronic LOAEL
- Skin irritancy and sensitization
- Eye irritancy
- Aerobic biodegradability
- Fathead minnow LC50
- Daphnia magna EC50
DELIVERING NEW SCIENCE
Molecular simulations are essential to modeling and understand-
ing complex biomolecular systems. The latest release of BIOVIA’s
predictive science application, Discovery Studio, includes anti-
body excipient interactions prediction to enhance biotherapeutics
formulations. Built on BIOVIA Pipeline Pilot™, Discovery Studio®
is uniquely positioned as the most comprehensive, collaborative
modeling and simulation application for Life Sciences discovery
research.
DISCOVERY STUDIO 2022
Part of the 2022 BIOVIA product release series, Discovery Studio
2022 continues to deliver scientific developments in the areas
of biotherapeutics, simulations, and small molecule research.
NEW AND ENHANCED SCIENCE
New! Excipient Interactions Prediction.
• A new protocol, Predict Excipient Interactions, predicts the
preferential interaction of common excipients with antibody
surface residues for antibody formulation1.
• Support Vector Machine and Elastic Net machine learning
models generated from molecular dynamics simulations
predict molecular interactions for 6 formulation excipients
– sorbitol, sucrose, trehalose, proline, arginine·HCl and NaCl.
• Protein surfaces colored by Γ23 values (preferential interaction
coefficients) show interactions of local excipient molecules
compared to local water molecules.
• Used with existing aggregation and viscosity prediction tools, this
enables formulation design earlier in the development process.
Figure 1: Positive values in red indicate higher Γ23 values,
corresponding to more local excipient molecules, while
negative values in blue correspond to more local water
molecule interactions with the antibody.
New! Feature Generation Components.
• Calculate Protein Features and Calculate Sequence Descriptors
components calculate structure- and sequence-based descrip-
tors for machine learning.
• New scripting APIs to support standard and specialized feature
calculations, e.g., a specific interatomic distance for a particular
residue type.
Enhanced! Protein modeling.
• Calculate Protein Formulation Properties protocol now auto-
matically creates aggregation sites and surfaces for analysis
with the View Aggregation Sites tools.
• Positively and negatively charged areas in the Charge Map are
available as Site groups.
• Substructure search for ligand query added to the RCSB Struc-
ture Search protocol, as well all available match types from the
RCSB server.
Enhanced! Simulation enhancements.
• Reduced the memory usage of large solvated systems when
running simulations.
• Assign Forcefield protocol now works with RNA and DNA
templates from custom RTF files.
Enhanced! Various pharmacophore modeling enhancements.
• Nucleic acids supported in the Interaction Pharmacophore
Generation protocol.
• Pharmacophores from a receptor-ligand complex that have
more than 50 non-bond interactions can be built with the
Interaction Pharmacophore Generation protocol.
Figure 2: View the details of aggregation sites.
Figure 3: Nucleic acid interaction pharmacophore.
PARTNER SCIENCE
• CHARMm: Incorporates the academic release CHARMM,
version c44b22.
• NAMD: Distributed with both CPU and GPU editions, version 2.13.
• MODELER: Incorporates the latest release of the academic
MODELLER, version 9.243.
• BLAST+: The BLAST+ version 2.10.1.
• GOLD: Supports GOLD 2021.
COMPATIBILITY
Discovery Studio 2022 is built on BIOVIA Pipeline Pilot 2022.
REFERENCES
1. Cloutier T. K., Sudrik C., Mody N., Sathish H. A., Trout B. L.,
Molecular Pharmaceutics, 2020, 17, 3589-3599.
2. Brooks B. R., Brooks III C. L., Mackerell A. D., Karplus M.,
et
al, J. Comp. Chem., 2009, 30, 1545-1615.
3. Eswar N., Marti-Renom M. A. Webb B., Madhusudhan M. S.,
Eramian D., Shen M., Pieper U., Sali A.,
Current Protocols in
Bioinformatics, John Wiley & Sons, Inc., 2006, Supplement
15, 5.6.1-5.6.30.
• Ligand Profiler protocol includes an option to maximize the
number of features or maximize the pharmacophore fit when
selecting the best fit.
• Screen Library protocol includes an option to report only the
highest fitting pharmacophore for each molecule.
Enhanced! Client functionality enhancements.
• Access the AlphaFold Protein Structure Database from the
Open URL dialog.
• Color proteins from the AlphaFold Protein Structure Database
based on the per-residue confidence score.
• Calculate RMSD and similarity properties without alignment.
• Analyze reports with sortable tables without an active server.
Figure 4: A protein from the AlphaFold Protein Structure
Database colored by the per-residue confidence score.
Related products
-

Leapfrog Geo 5.0.1 license software
$ 140.00 Add to cart Quick View -

SAGE Profile 3.0.26 cracked software
$ 190.00 Add to cart Quick View -

STEAG EBSILON 12 Professional cracked version
$ 130.00 Add to cart Quick View -

CADMATIC 2016Q1 (Diagram, Hull, Outfitting, Plant, Design) cracked
$ 130.00 Add to cart Quick View -

Geosoft 9.5 licensed software and cracked
$ 165.00 Add to cart Quick View -

DATAMINE 2018 full suite – cracked license
$ 180.00 Add to cart Quick View -

ZOND software fully cracked
$ 250.00 Add to cart Quick View -

Hydro GeoAnalyst 8.0 build 17.18.0925.1 cracked version
$ 220.00 Add to cart Quick View -

GRLWEAP 2010-6 software already crack
$ 140.00 Add to cart Quick View -

EMTP-RV V3.4 cracked version
$ 150.00 Add to cart Quick View